Research Topics
1. Bacteriophage inhibition of antibiotic-resistant pathogenic microbes and finding novel therapeutic strategies (PHARMS)
Antimicrobial resistance (AMR) is a major threat to global health, global economies, and humanity itself. Worldwide, AMR has killed more than 700,000 people, including over 25,000 cases in Europe every year. AMR is particularly deadly where malnutrition and co-infections are widespread, especially in developing countries. The rapid spread of AMR, and its devastating consequences for patients as well as healthy individuals, makes it one of the most important scientific challenges of our time. Research on novel AMR control strategies is clearly needed, despite the significant progress made in searching for new antibiotics. Viruses of bacteria, bacteriophages, are the nature’s most prevalent bacterial predators, which can be employed to fight AMR as a complement to antibiotic therapy. Coupling the bactericidal effects to host-recognition machineries, promising progress has been made in treating AMR bacteria using: i) lytic phages which can directly lyse hosts or a cocktail of different lytic phages to overcome bacterial resistance; ii) phage-encoded bactericidal peptides or enzymes, such as endolysins (lysins), which are peptidoglycan hydrolases involved in cell lysis during phage replication. To avoid adverse reactions caused by immune recognition of phages, phages with innate characteristics that are unlikely to elicit an immune response or mutant phages that are not recognized by the immune system are preferred for medical treatment. PHARMS will focus on three AMR bacteria: A. baumannii, H. influenzae, and H. pylori, all of which are major human pathogens in this year’s WHO guideline of twelve AMR bacterial families prioritized for R&D efforts. Three synergistic work packages are tailored for PHARMS: WP1 is designed to understand the infection strategies of phages in a high-throughput, cultivation-independent manner, and to provide information that guides targeted phage isolations; WP2 aims to targeted isolate representative divergent phages and understand the phage bactericide systematically in achieved divergent phage isolates, in order to engineer a new series of phage vectors with superior bactericidal potential by employing yeast-based engineering of phages; WP3 is designed to provide direct molecular confirmation for the function and mechanisms of phage-encoded bactericidal peptides and enzymes in selected phage isolates. In addition, in the BMBF founded project COVPHA (Interrogating COVID-19, the lung microbiota, and therapeutic phages to mitigate secondary lung infection and inflammation / Untersuchung von COVID-19, der Lungenmikrobiota und therapeutischer Phagen zur Linderung von sekundären Lungeninfektionen und –entzündungen), we aim to combat those AMRs associated with bacterial pneumonia of COVID-19 patients. The ongoing COVID-19 pandemic highlights the need to understand the complex viral-bacterial interactions in human health.
2. Functional roles of viruses in the natural environment and human-associated microenvironment
2.1 Impact of viruses in human (/animal) health and disease
The human microbiome is a unique polymicrobial community protecting the human host on the one hand from pathogenic assault by competing for sites of attachments and nutrients, as well as producing antimicrobial substances, and on the other hand altering the host’s metabolism by modulating lipid metabolism and glucose homeostasis, absorbing lipid-soluble vitamins, or activate respectively inactivate drugs. The composition of the microbiota thus affects metabolic, regulatory and morphogenetic networks. Members of the virome influence the phenotype of the host in a combinatorial manner by interacting with other members of the microbiome (such as other members of the virome itself, the bacterial microbiome or the mycobiome) and by interacting with individual variations in host genetics. Together these interactions may influence a range of phenotypes, which may be important for health and disease. We want to elucidate the functional roles of viruses especially with regard to disease progression. The method of choice for these questions is the application of metagenomic sequencing of a broad range of human samples deriving from healthy and diseased patients. In a further step, animal models are utilized to investigate the functional impact of exclusive enteral nutrition via microbiome and phageome changes in intestinal inflammation and cancer (DFG CRC/SFB 1371, Microbiome Signatures - Functional Relevance in the Digestive Tract).
2.2 Impact of viruses on microbial-driven contaminant biodegradation
Contamination with organic pollutants, such as petroleum hydrocarbons, is widespread in groundwater and a notorious threat to our water resources. Biodegradation is the single most important and sustainable process for contaminant breakdown. The biological activity that governed by numerous processes of which the current perspective is only evolving hydrology, biogeochemistry and environmental microbiology. One important piece of the puzzle, however, has been rarely touched: viruses. This project aims to elaborate a ground-breaking new perspective, the viral-driven degradation. Viruses are ubiquitous and abundant global players that impact microbial communities through mortality and horizontal gene transfer to modulate microbial metabolism. During the past years diverse phenomena critical to the biology of microbes have been described to be driven by viruses, especially with respect to rapid environmental changes. We hypothesize that degradation of contaminants is greatly impacted by viruses through (i) horizontally transfer host metabolic genes related to contaminant degradation, and (ii) specifically lysing key bacterial degraders. In a cutting-edge and interdisciplinary research endeavor, we want verify our hypotheses using model aerobic and anaerobic degraders for the group of benzene, toluene, ethylbenzene and xylenes (BTEX) and polycyclic aromatic hydrocarbons (PAHs). Pillaring on recent advances in methodology, the culture-independent, high-throughput approach “Viral-Tagging” which allows to link natural viruses to their hosts, and vice versa, carefully designed field surveys and microcosm experiments as well as metagenomics sequencing will be conducted.
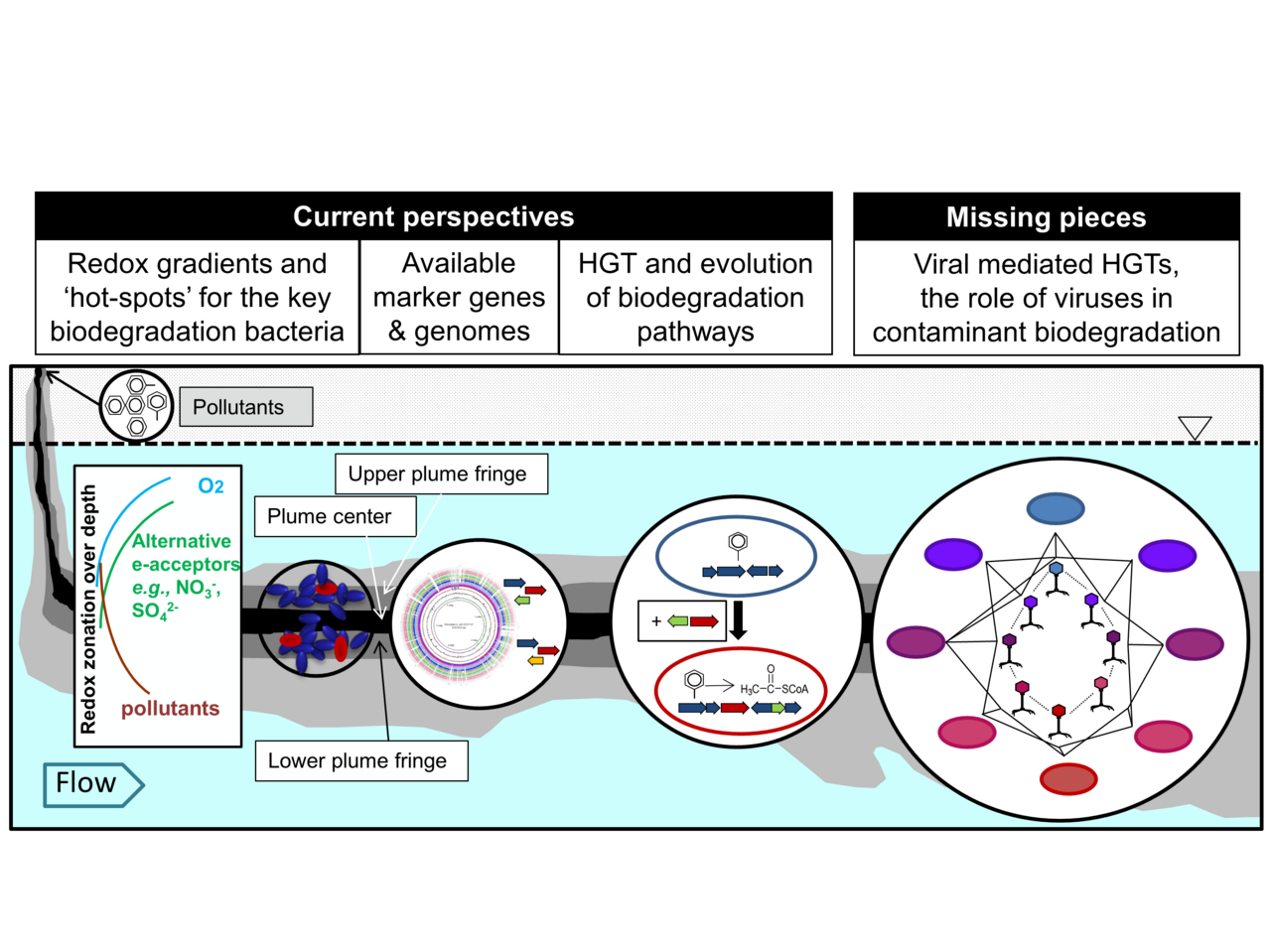
Figure 3. Current paradigms and missing pieces of contaminant groundwater ecology (HGT=horizontal gene transfer)
COMPUTATIONAL PREDICTION OF VIRUS-HOST INTERACTIONS IN THE MICROBIOME
Despite large ongoing efforts to identify viruses in humans, animals and the environment, there are still multiple challenges remaining in the identification of novel viruses, as well as their host. These challenges remain both in the wet-lab as well as on the bioinformatical side. On the bioinformatical side, the most frequently used similarity-based virus identification approaches are largely limited by available databases, which are incomplete and often poorly annotated. We aim to set up a multiple layer database for phages and their hosts in the frame work of the European Virus Bioinformatics Center and an EU Innovative Training Networks (ITN) project (Marie Skłodowska Curie Action). Existing resources and genomic signals will be leveraged to infer missing/unobserved and probable hosts of phages, e.g., where infection has not been observed due to host immunity. Machine learning models will be trained on this data to predict phage-bacteria interactions at the species and strain levels, with the ultimate goal of host prediction in microbiome.